
Philipp

Fabian
Rückblick
- Prozess: Satz von in Wechselbeziehung oder Wechselwirkung stehenden Aktivitäten, der Eingaben in Ergebnisse umwandelt
- Aktivität: Satz von einer oder mehreren aufeinander bezogenen oder sich gegenseitig beeinflussenden Aufgaben
- Aufgabe: eine Teilarbeit, die erledigt werden muss
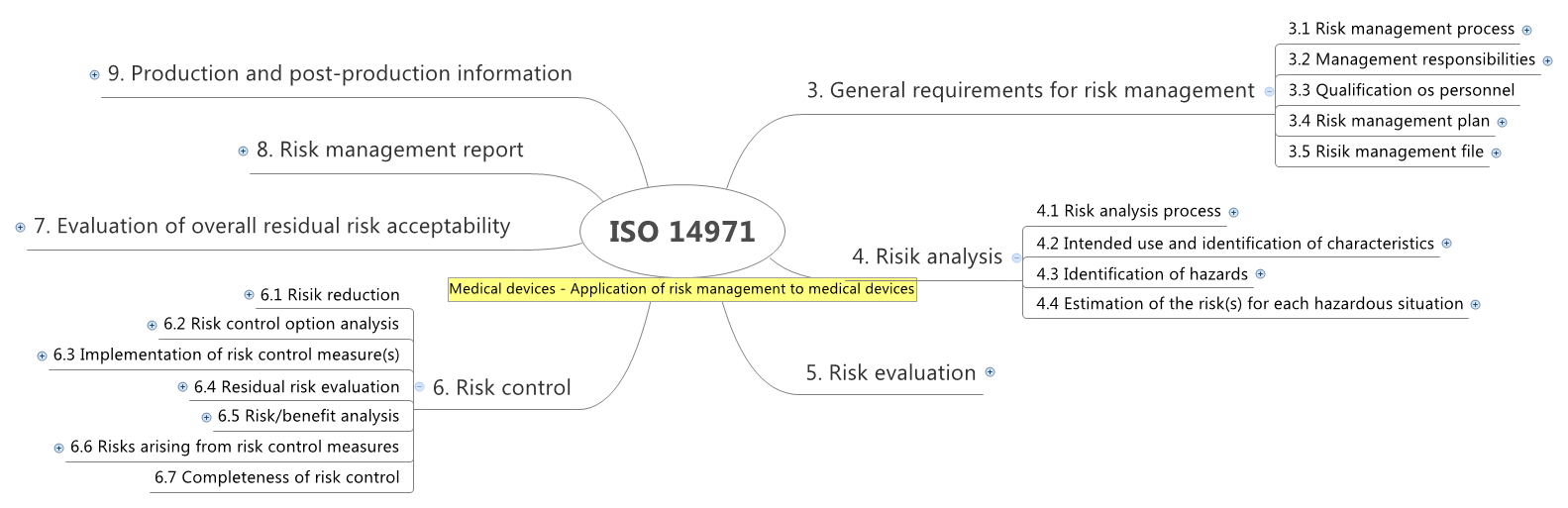
Prozesse der IEC 62304
- Software Entwicklungsprozess
- Software Wartungsprozess
- Software Risikomangement Prozess
- Software Problemlösungsprozess
- Software Konfigurationsmanagement Prozess
Software Entwicklungsprozess – Planung
Erste Teilaktivität des Software Entwicklungsprozesses
Planung – Allgemein
Aktivität: Planung der Softwareentwicklung
Output: Software Entwicklungsplan
- Rollen und Verantwortlichkeiten
- Software Entwicklungsmodell
- Projektphasen
- Outputs in den Phasen
- Review-Kriterien der Outputs
- Projektphasen
- Verweis auf weitere Prozesse der IEC 62304
- Software Entwicklungsprozess im Projekt
- Allgemein
- Tooling
- Compiler, Interpreter, Entwicklungsumgebung, Versionskontrollsystem…
- Coding Guidelines
- Tooling
- Allgemein
- Planung der Software Anforderungen
- Planung der Software Architektur und des Detail Design
- Planung der Implementierung
- Planung der Software Unit Verifikation
- Planung der Software Integration
- Planung der Software System Verifikation
- Planung des Software Release
Planung – Softwareanforderungen
- Wo werden die Softwareanforderungen erfasst?
- Word, Excel, IBM Doors, Redmine, Polarion, OpenProject, CodeBeamer, HelixALM, JIRA, Greenlight guru GO, …
- Evtl. Satzschablonen verwenden
- Use Cases oder “Requirement-Schablone” (shall, should, may)
- Sicherstellen der Traceability
- von den Systemanforderungen zur Architektur und Verifikation
- Festlegen des zu unterstützendes Betriebssystems
- Festlegen der zu unterstützende Hardware
- Sicherstellen der Verifizierbarkeit
- Sicherstellen, dass die Softwareanforderungen widerspruchsfrei und vollständig sind
- Review und Freigabeprozess
- Granularitär / Detailierungsgrad der Anforderungen festlegen
- Wertebereiche, Datentypen und Reaktionen des Systems (auch bei Fehleingaben)
- Schnittstellen zu anderen Systemen
- Reaktionszeiten
- Interoperabilität
- Datenvolumen und Last auf dem System
Planung – Architektur und Detailliertes Design
- Wo wird die Software Architektur erfasst?
- Definition Software-System, -Item und -Unit
- Beschreiben was für Software Item und Units dokumentiert werden muss
- Static View
- Dynamic View
- Interfaces
Planung – Implementierung
- Commit Workflows
- Anwendung der Tools
Planung – Software Unit Verifikation
- Planung der Verifikation der Schnittstellen von Software-Unit
- Statische Code-Analyse
- Code Reviews
- Dynamische Analyse
- Beschreiben in welchem System die Software Units verifiziert wird
- Akzeptanzkriterien festlegen (beim Verhalten der Schnittstellen)
- Testabdeckung festlegen
Planung – Software Integration
- Integration Strategy (Top-down, bottom-up, …)
- Beschreiben in welchem System die Integration und Integrationstest durchgeführt wird
- Festlegen was in den Testplan und -Bericht kommt
- Akzeptanzkriterien festlegen
- Zuverlässigkeit und Robustheit
- Performance
- Kompatibilität und Wartbarkeit
- Festlegen wie Anomalien in den Problemlösungs-Prozess einfließen
Planung – Software System Verifikation
- Beschreiben in welchem System die Software System Verifikation durchgeführt wird
- Sicherstellen, dass alle Software-Anforderungen verifiziert werden
- Festlegen was in den Testplan und -Bericht kommt
- Akzeptanzkriterien festlegen
- Festlegen wie Anomalien in den Problemlösungs-Prozess einfließen
Planung – Software Release
- Beschreiben in welchem System das Software Release durchgeführt wird
- Festlegen wo die Software-Releases abgelegt werden
- Festlegen wo offenen Anomalien und Bugs dokumentiert und bewertet werden
- Schema der Software-Versionsnummern festlegen
- Sicherstellen, dass alle zuvor genannten Aktivitäten durchgeführt wurden
- Mittels Checkliste durch die technische Dokumentation gehen und Review durchführen
Allgemeine Software Entwicklungsmodelle
Vorgehensmodelle
- Es gibt keine Vorgabe in der Norm IEC 62304
- Forderung: Betrachtung der Abhängigkeiten zwischen den Aktivitäten
- Erst die Anforderungen und die Software Archtektur abschließen, dann die Risikoanalyse abschließen.
- Mehrere Vorgehensmodell sind denkbar.
- Die IEC 62304 zählt u.a Wasserfallmodell, iterativ-inkrementell, evolutionär etc. auf.
- Entwicklungsmodelle können textuell oder als Flowchart beschrieben werden.
Wasserfallmodell
- Alle Entwicklungsschritte des Wasserfallmodells werden nacheinander durchgeführt.
- Phasenbasiert, erst eine Phase abschließen, dann Beginn der nächsten Phase
- Review der Ergebnisse zwischen den Phasen
-
Vorteile
- Einfach.
- Phasenabschlüsse geben Sicherheit über den Projektfortschritt.
- Klare Abschätzung von Kosten und Umfang bei stabilen Anforderungen.
- Nachteile
- Fehler werden meistens erst spät entdeckt.
- Wiederholung von Phasen im Fehlerfall.
- Unflexibel gegenüber Änderungen im Projekt.
V-Modell
- Weiterentwicklung des Wasserfallmodells
- Jetzt: Jede Phase hat eine gegenüberliegende Testphase
- Vorteile
- Testaspekte können früher betrachtet werden.
- Fehler in der Spezifikation oder im Design können früher gefunden werden.
- Durch Berücksichtigung von Testaspekten ist die Dokumentation besser.
- Nachteile
- Implementierungsfehler werden erst beim Testen entdeckt, meistens sehr spät.
- Auch hier: Im Fehlerfall müssen Phasen wiederholt werden, was sehr zeitaufwändig ist.
Iterativ-inkrementelle Modelle
- Iterativ: Einzelne Phasen werden direkt durchlaufen
- Inkrementell: Pro Durchlauf wird der Funktionsumfang erweitert
- Fehler, die am Ende jedes Phasendurchlaufs entdeckt werden, werden im nächsten Durchlauf behoben.
Agiles Entwicklungsmodell
- Die Basis agiler Softwareentwicklung ist das iterativ-inkrementelle Modell.
- Ziel ist es, bürokratische Strukturen abzubauen und den Fokus auf die kundennahe Entwicklung zu richten.
- Weitverbreitet ist dabei Scrum.
- Mit der IEC 62304 ist dieses Modell zwar vereinbar, wird allerdings noch etwas zögerlich eingesetzt, da die Norm sehr V-Modell-artig aufgebaut ist. Viele schrecken vor der Kombination aus agiler Welt und V-Modell-Welt noch zurück.
Quellen
SW Entwicklungsplanung:
- IEC 62304:2015
SW Entwicklungsmodelle:
- https://de.wikipedia.org/wiki/Wasserfallmodell
- https://de.wikipedia.org/wiki/V-Modell
- https://de.wikipedia.org/wiki/Inkrementelles_Vorgehensmodell
- https://de.wikipedia.org/wiki/Scrum
- SW Entwicklung für Medizinprodukte – SW-Lebenszyklus-Prozesse; Prof. Dr. Michael Munz, HS Ulm









{kind=link}