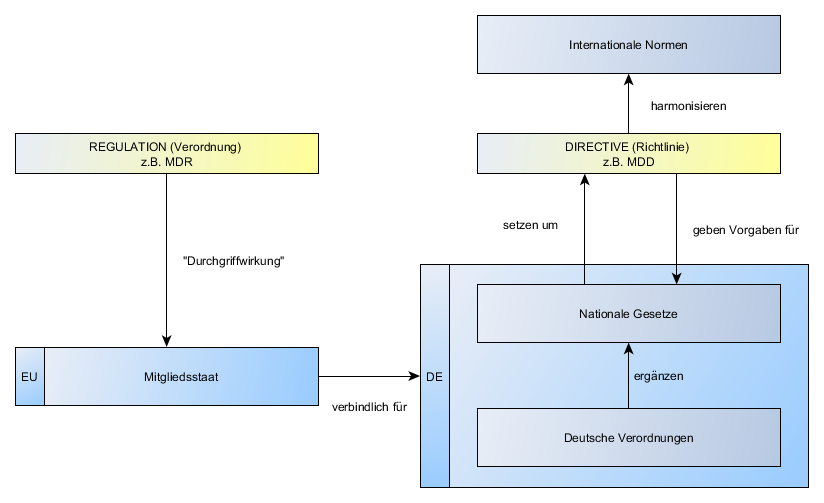
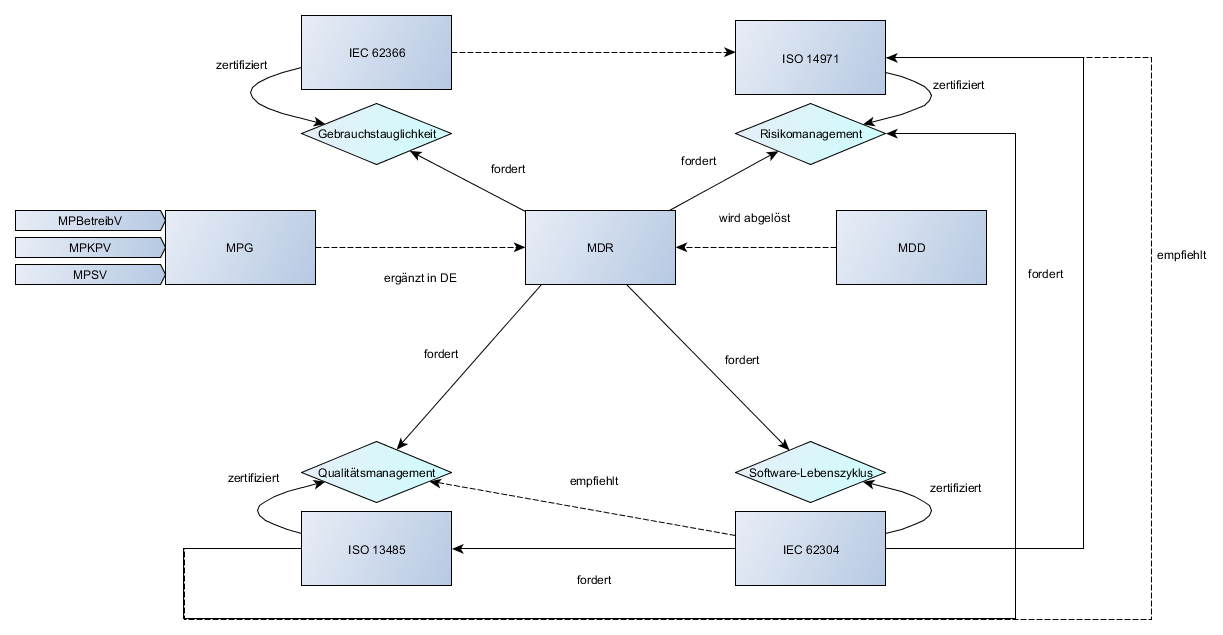
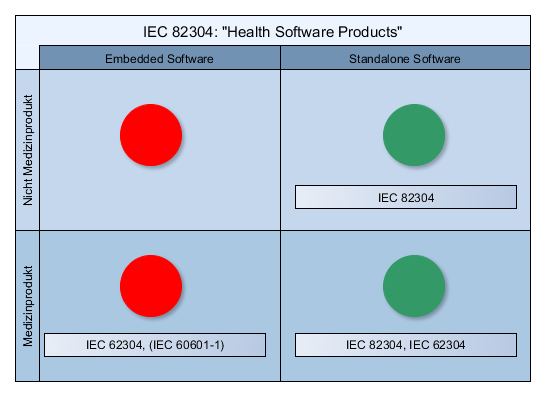
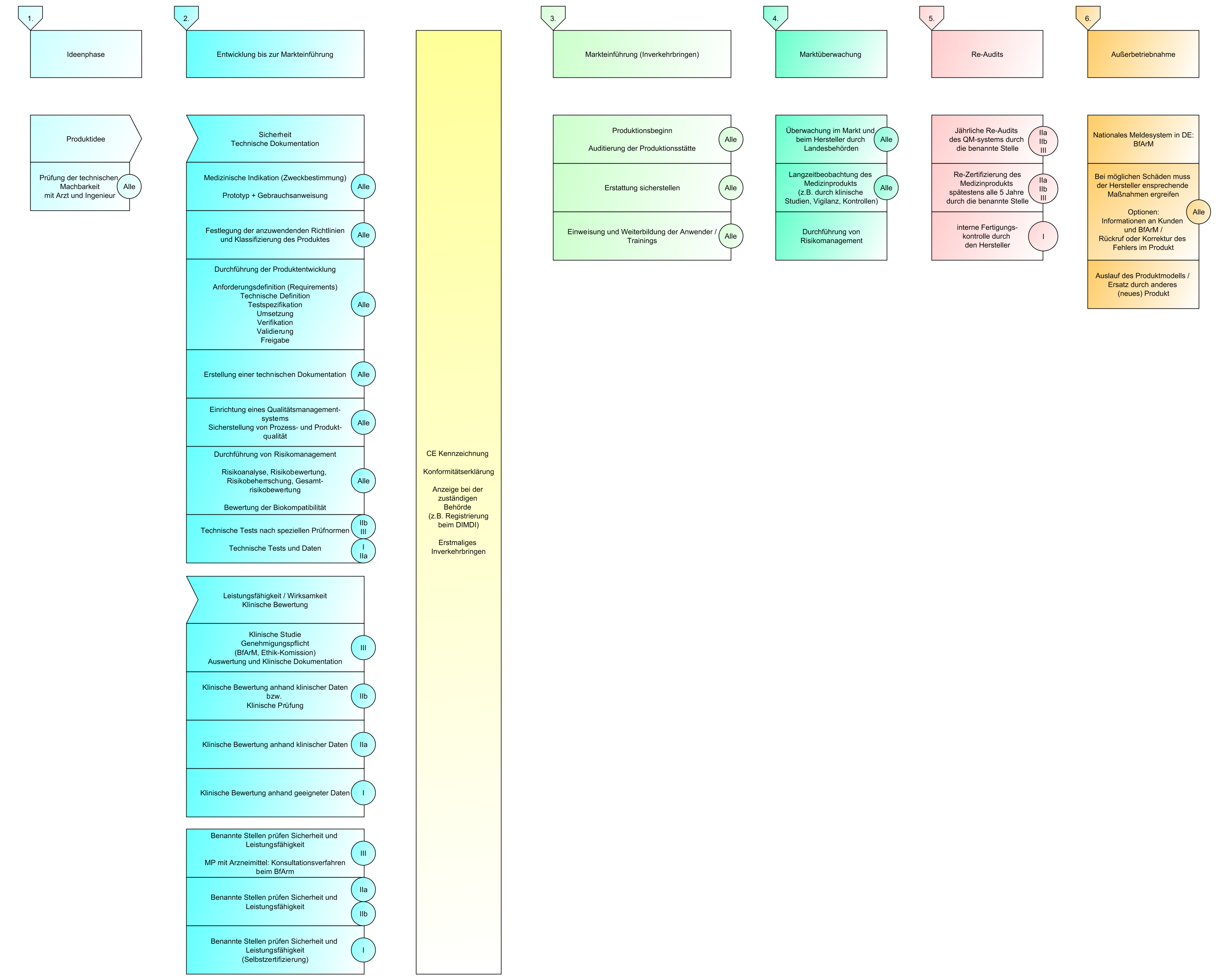
Lebenszyklus eines Medizinprodukts
Zweckbestimmung und bestimmungsgemäßer Gebrauch
Jeder Hersteller sollte mit der Zweckbestimmung beginnen. Das ist quasi das wichtigste Dokument am Anfang einer Produktidee im Medizinbereich.
Die Zweckbestimmung entscheidet, ob es sich bei dem Produkt um ein Medizinprodukt handelt oder nicht. Damit entscheidet der Hersteller ob es ein Medizinprodukt ist oder nicht. Die Aufsichtsbehörden haben natürlich das letzte Wort. Das Bundesinstitut für Arzneimittel und Medizinprodukte ist Zuständig für die Abgrenzung und Klassifizierung von Medizinprodukten.
Das Messen von beispielsweise Blutdruck und Puls ist eine technische Funktion. Erst die Zweckbestimmung macht das Produkt zum Medizinprodukt (oder eben auch nicht).
Inhalt der Zweckbestimmung
Eine Zweckbestimmung sollte die folgenden Aspekte dokumentieren:
- wie das Produkt der Diagnose, Therapie oder Überwachung von Krankheiten oder Verletzungen oder physiologischer oder anatomischer Parameter dient. Das kann ein physikalisches Funktionsprinzip (Quasi die Behandlung: Strahlung, …), Dokumentation, Berichterstellung, Algorithmen, Datenverarbeitung oder Planung sein. Auch die Anwendungsort (Körperteil am Patienten) und die Anwendungsdauer sind hier zu dokumentieren
- die vorgesehene Patientengruppe mit Informationen wie Alter, Geschlecht, Gesundheitszustand, Beeinträchtigungen, Krankheiten, familiäres Umfeld und Gewicht,
- welche Körperregion bzw. welches Gewebe untersucht, diagnostiziert, therapiert oder überwacht werden soll,
- die Benutzergruppen einschließlich Alter, Beruf, Ausbildung/Weiterbildung, Geschlecht, Sprachkenntnisse, spezielle relevante Fähigkeiten oder Erfahrungen mit bestimmten Produkten. Hier ist es auch sinnvoll die Länder zu nennen, in denen das Produkt eingesetzt werden soll.
- den Benutzungskontext, also Kernaufgaben, Häufigkeit der Anwendung, Arbeitsbelastung, zu erzielende Ergebnisse, sowie
- den Ort der Anwendung und dort vorherrschende Verhältnisse wie Helligkeit, Temperatur, Luftfeuchtigkeit, Lautstärke in der Umgebung, Sichtweite und die mentale Belastung
- Erste Abschätzung der Marktdaten wie geplante Lebensdauer des Produktes, Anzahl der zu verkauften Produkte, Anzahl der Anwendungen des Produktes pro Tag und pro Patient
Die Fragen im Anhang C in der ISO 14971 lassen sich sehr gut als Checkliste hernehmen um sicherzustellen, dass alle Aspekte die eine Zweckbestimmung beinhalten sollte tatsächlich berücksichtigt wurden. Das Inhaltsverzeichnis der Norm ist öffentlich einsehbar. Die Fragen sind als Unterkapitel in der Norm beschrieben. Damit sind die Fragen für Jeden öffentlich einsehbar:
- https://www.iso.org/obp/ui/#iso:std:38193:en
- https://www.beuth.de/de/norm/din-en-iso-14971/170088031
Bestimmungsgemäßer Gebrauch
Der bestimmungsgemäßen Gebrauchs enthält zusätzlich zur Zweckbestimmung noch weitere Informationen zum Produktlebenszyklus (von der Installation über Instandhaltung, Transport und Lagerung bis zur Entsorgung wie z.B.:)
- Umweltbedingungen für Lagerung und Transport (Temperatur, Luftfeuchtigkeit, Druck)
- Installationsbedingungen (z.B. Installation durch Fachpersonal)
- Wartungsbedingungen (Intervalle) oder Fernwartung
- Reinigung
Klassifizierung von Medizinprodukten und Konformitätsbewertungsverfahren
In der letzten Sendung haben wir über die Konformitätsbewertungsverfahren geredet und für welche Klassen, welche Verfahren angewendet werden dürfen. Heute gehen wir noch mal einen Schritt zurück und schauen uns die Klassen einmal genauer an.
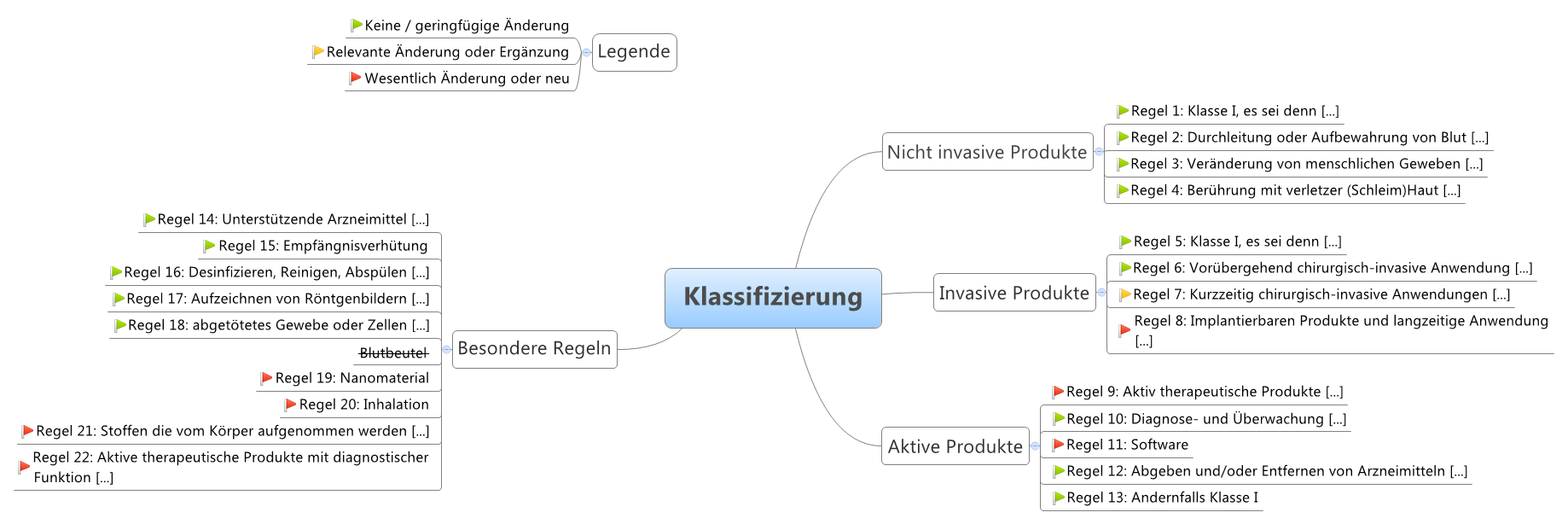
Es gibt vier Klassen von Medizinprodukten:
| Klasse | Risiko | Beschreibung | Beispiele |
|---|---|---|---|
| III | Sehr hohes Risiko | zur langfristigen Medikamentenabgabe, Inhaltsstoff tierischen Ursprungs und im Körper, unmittelbare Anwendung an Herz, zentralem Kreislaufsystem oder zentralem Nervensystem, und invasive Empfängnisverhütung | Hüft-und Kniegelenkimplantate, Herzkatheter, Brustimplantate, ... |
| IIb | Hohes Risiko | systemische Wirkungen, Langzeitanwendungen, nicht invasive Empfängnisverhütung, langzeitig ≥ 30 Tage, sonst wie bei kurzzeitig | Intraokularlinsen, Kondome, Röntgengeräte, Infusionspumpen, ... |
| IIa | Mittleres Risiko | mäßiger Invasivitätsgrad, kurzzeitige Anwendungen im Körper (im Auge, intestinal, in chirurgisch geschaffenen Körperöffnungen) kurzzeitig ≤ 30 Tage, ununterbrochen oder wiederholter Einsatz des gleichen Produktes | Zahnfüllungen, Röntgenfilme, Hörgeräte, Ultraschallgeräte, ... |
| I | Geringes Risiko | geringer Invasivitätsgrad, kein oder unkritischer Hautkontakt, vorübergehende Anwendung ≤ 60 Minuten | Lesebrillen, Rollstühle, Mullbinden, Fieberthermometer, ... |
- Messfunktion (Im)
- steril (ls)
- wiederverwendbare chirurgische Instrumente (lr)
Die Klassen dienen der Einteilung der Produkte nach der “Verletzbarkeit des menschlichen Körpers”. Diese wiederum definiert sich über die Zweckbestimmung des Herstellers.
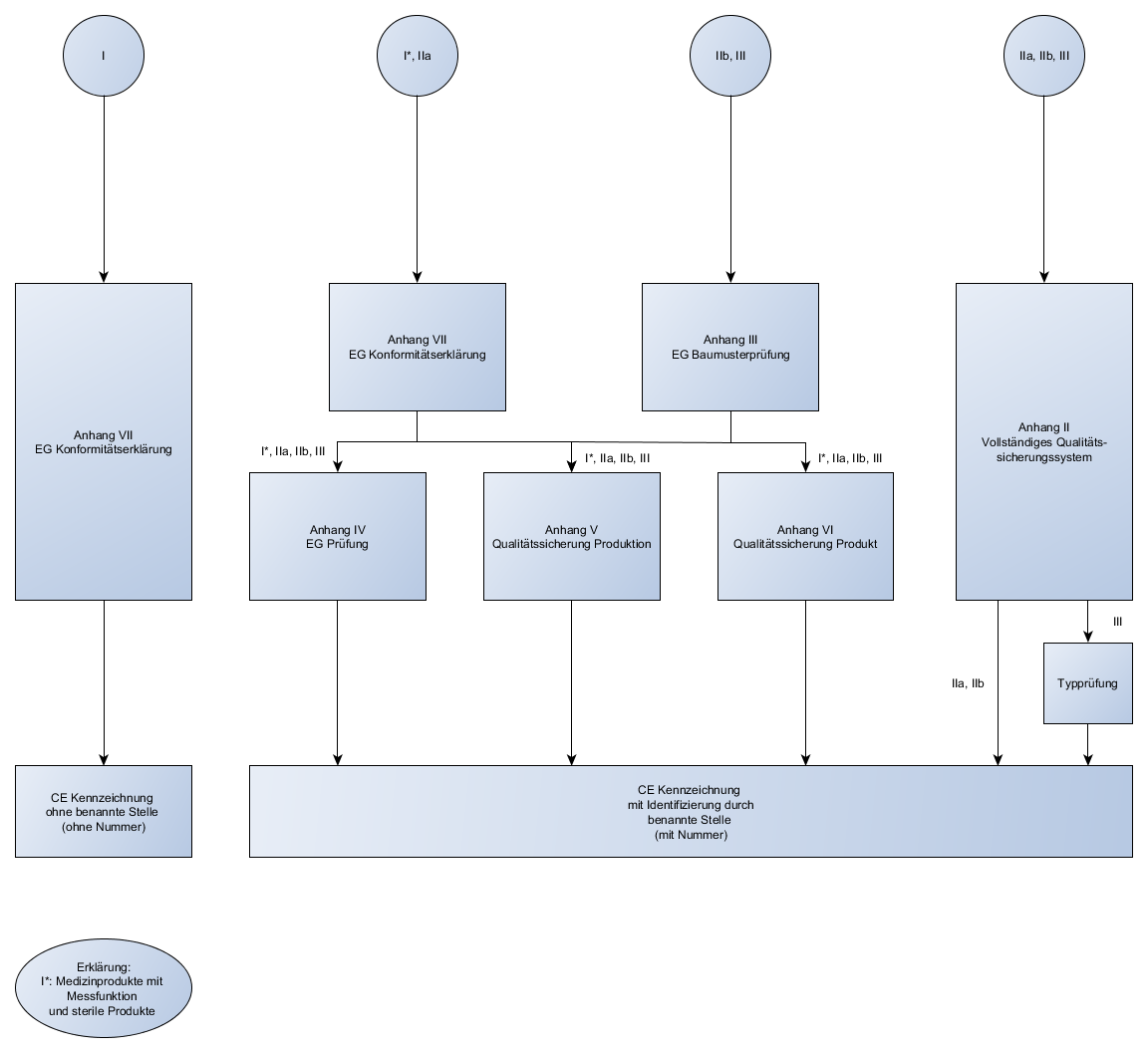
Abhängig von dieser Klassifizierung bestimmen sich die Konformitätsbewertungsverfahren, die für das Produkt angewendet werden dürfen.
Nicht zu verwechseln mit: den Softwaresicherheitsklassen gemäß IEC 62304!
Konformitätsbewertungsverfahren:
- Die verschiedenen Konformitätsbewertungsverfahren wurden in der letzten Sendung behandelt
- Die verschiedenen Regeln zur Klassifizierung wurden in der letzen Sendung behandelt
- MDD ist “einfach” und verstanden
- MDR ist noch neu und es fehlt an Erfahrungswerten
Quellen:
Klinische Bewertung
Anhang XIV MDR
- Eine klinische Bewertung ist für jedes Medizinprodukt ein zwingend erforderlicher Teil des Konformitätsbewertungsverfahrens (Anhang X der MDD).
- Kann im einfachsten Fall auf vorhandener Literatur basieren, wenn nachgewiesener Weise die Daten für die technischen Anforderungen, als auch für die Zweckbestimmung eines vergleichbaren Produktes erhoben wurden.
- Falls nicht, muss eine klinische Prüfung durchgeführt werden (strengere Reglementierungen)
- Jede klinische Prüfung muss beim Deutschen Institut für medizinische Dokumentation und Information (DIMDI) beantragt und von einer Ethik-Kommission [MPKPV] genehmigt werden.
Konformitätserklärung und CE Kennzeichnung
Der Hersteller erklärt die Konformität
Die Konformitätserklärung (Declaration of Conformity) muss in der Regel unter Einbezugnahme einer benannten Stelle erfolgen.
Ein Hersteller darf nur Produkte in Verkehr bringen die explizit auf der Konformitätserklärung genannt sind.
Das Kürzel der benannte Stelle ist mit auf dem CE-Kennzeichen eines jeden Produktes vorhanden. Außer bei Klasse I.
Das CE-Kennzeichen an sich wurde in der letzten Folge über die MDR erklärt.
Inverkehrbringen
Erst mit der CE-Kennzeichnung und nach erfolgter Registrierung ist das Medizinprodukt für den europäischen Binnenmarkt freigegeben und darf dann ohne Einschränkungen gehandelt werden.
- Vorher ist eine Verbreitung untersagt!
- Im Rahmen der Produktvalidierung muss der Hersteller darauf achten, dass die Bereitstellung zur Validierung nicht bereits einem Inverkehrbringen entspricht.
- Definition: Inverkehrbringen (MDD): „Inverkehrbringen: erste entgeltliche oder unentgeltliche Überlassung eines Produkts, das nicht für klinische Prüfungen bestimmt ist, im Hinblick auf den Vertrieb und/oder seine Verwendung innerhalb der Gemeinschaft, […].“
- Jedes Medizinprodukt muss bei der DIMDI registriert werden.
Überwachung nach dem Inverkehrbringen / Post-Market Surveillance (MDR)
- Mit der Markteinführung des Produktes endet die regulatorische Pflicht des Herstellers nicht
- Vielmehr muss während des gesamten Lebenszyklus des Produktes der Markt beobachtet werden
- Der Lebenszyklus eines (Medizin-)Produkts endet dann, wenn die vorgesehene Lebensdauer des zuletzt ausgelieferten Produkts abgelaufen ist oder wenn das zuletzt im Markt befindliche Produkt im Markt außer Betrieb genommen und entsorgt wurde
- Dies fordert die MDR, als auch Normen des QM und Risikomanagements
- Die ISO 13485 fordert, dass die Hersteller die Wirksamkeit Ihres QM-Systems und die Sicherheit Ihrer Medizinprodukte durch Marktbeobachtung sicherzustellen.
- Die ISO 14971 fordert, dass die Hersteller nach in-verkehr bringen Ihrer Medizinprodukte feststellen ob die Wahrscheinlichkeiten und Schweregrade richtig angenommen wurden. Es muss auch kontinuierlich geprüft werden, dass die daraus resultierenden Risiken und Risikoakzeptanzkriterien immer noch gültig sind.
Definition aus der MDR: „Überwachung nach dem Inverkehrbringen“ bezeichnet alle Tätigkeiten, die Hersteller in Zusammenarbeit mit anderen Wirtschaftsakteuren durchführen, um ein Verfahren zur proaktiven Erhebung und Überprüfung von Erfahrungen, die mit den von ihnen in Verkehr gebrachten, auf dem Markt bereitgestellten oder in Betrieb genommenen Produkten gewonnen werden, einzurichten und auf dem neuesten Stand zu halten, mit dem ein etwaiger Bedarf an unverzüglich zu ergreifenden Korrektur- oder Präventivmaßnahmen festgestellt werden kann;
Das Kapitel VII(7) der MDR widmet sich diesem Thema https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:32017R0745&from=EN#L_2017117DE.01000101-d-009
Der Anhang III regelt die technische Dokumentation bezogen auf die Überwachung nach dem Inverkehrbringen https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:32017R0745&from=EN#d1e32-112-1
Quellen:
- https://medizintechnologie.de/innovation-im-fokus/expertenwissen/post-market-surveillance-pms/
- https://www.bvmed.de/de/recht/marktueberwachung
- https://www.johner-institut.de/blog/regulatory-affairs/post-market-surveillance/
Zwischenfälle
Jeder Hersteller muss sicherstellen, dass er Rückmeldungen von Kunden erhält, diese bewertet und ggf. Reaktionen in die Wege leiten kann.
- Hersteller müssen proaktiv im Markt befindliche, ähnliche Produkte, auf aufgetretene Probleme hin beobachtet.
- Alle Mitgliedsstaaten müssen ein nationales Meldesystem installieren, an das Hersteller, Betreiber und Patienten Vorfälle melden können.
- In Deutschland ist dies das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), das Formulare und Informationen bereithält.
- Bei Korrektur eines vom Hersteller im Markt befindlichen Produkts sind u.a. folgende Optionen an Maßnahmen möglich:
- Information an Kunden und BfArM (Swissmedic, FDA, …)
- Rückruf aller betroffenen Produkte (falls notwendig)
- Korrektur des Fehlers für kommende Produkte
BfArm Meldung über sich selbst entzündene OP-Handschuhe: https://www.bfarm.de/SharedDocs/Kundeninfos/DE/02/2013/04840-13_kundeninfo_de.html?submit=Filtern&oneOfTheseWords=fire+glove
Überwachung von Herstellern
- Anzeigepflicht vor Aufnahme der Tätigkeit: Laut MPG §25 „Allgemeine Anzeigepflicht“: „(1) Wer als Verantwortlicher […] seinen Sitz in Deutschland hat und Medizinprodukte […] erstmalig in den Verkehr bringt, hat dies vor Aufnahme der Tätigkeit unter Angabe seiner Anschrift der zuständigen Behörden anzuzeigen; […]“
- „zuständige Behörden“: Die Behörden sind den Ländern untergeordnet. Die Länder haben die Zuständigkeit unterschiedlich umgesetzt in z.B. Regierungspräsidien, Bezirksregierungen, Landesämtern, Landesdirektionen oder Gewerbeaufsichtsämter
- „DIMDI“: Beim DIMDI muss der Hersteller sich unter Angabe der Behörde sowie des Sicherheitsbeauftragten anmelden und das DIMDI leitet diese Anmeldung weiter an die Behörde. Die Behörde vergibt daraufhin eine Registrierungsnummer für den Hersteller.
vollständige Liste: Deutsches Institut für Medizinische Dokumentation und Information (DIMDI)
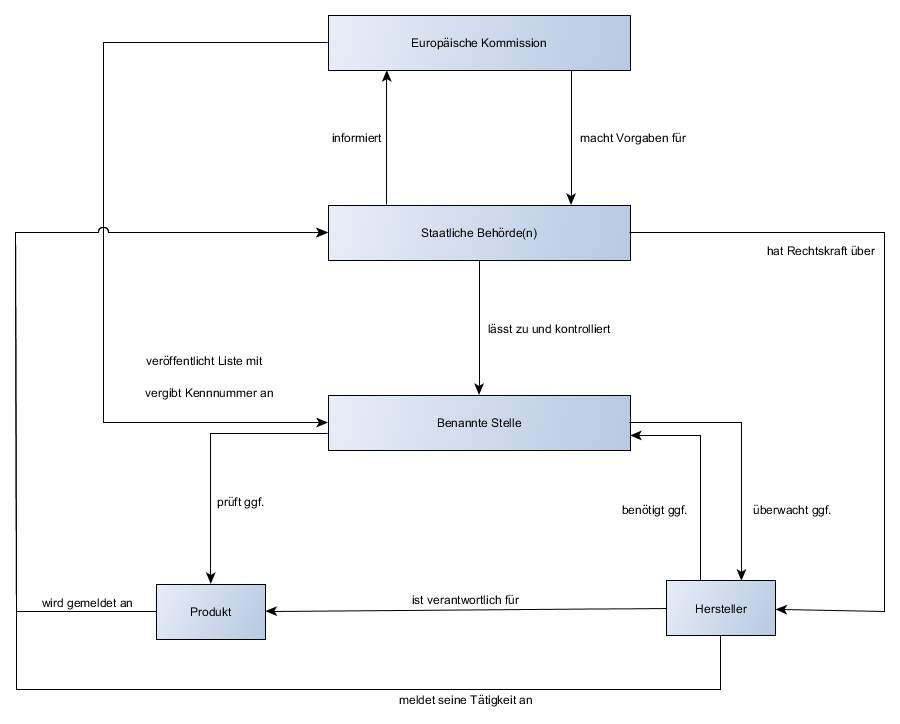
Überwachung von benannten Stellen
- Benannte Stelle: Bei höher klassifizierten Produkten als Klasse I, die nicht steril oder eine Messfunktion beinhalten, muss der Hersteller eine benannte Stelle einschalten. Diese ist ein privatwirtschaftlicher Dienstleister, die selbst keine Rechtskraft gegenüber dem Hersteller hat, allerdings selbst unter staatlicher Überwachung steht. Vorteile: Sicherheit der Produkte gewährleisten; fairer Wettbewerb in der EU
- Zulassung und Kontrolle der benannten Stelle obliegt den Behörden des Mitgliedstaates.
- Die Europäische Kommission macht Vorgaben an die Mitgliedstaaten, niemals aber selbst an die Hersteller. Sie verwaltet und veröffentlicht die von den Behörden gemeldeten Stellen und teilt Ihnen Kennnummern zu. Damit sind die Kennnummern, die der CE-Kennzeichnung bei Beteiligung einer benannten Stelle hinzugefügt werden müssen, europaweit eindeutig.
- EU Akkreditierung: (seit 1.1.2010) Nationale Akkreditierungsstelle wickelt Akkreditierung und Marktüberwachung, sowie Vermarktung von Produkten ab. In DE: Deutsche Akkreditierungsstelle (DAkkS)
weitere Quelle:
- Christian Johner , Matthias Hölzer-Klüpfel , Sven Wittorf: Basiswissen Medizinische Software – Aus- und Weiterbildung zum Certified Professional for Medical Software, Auflage: 2, dpunkt.verlag, 2015