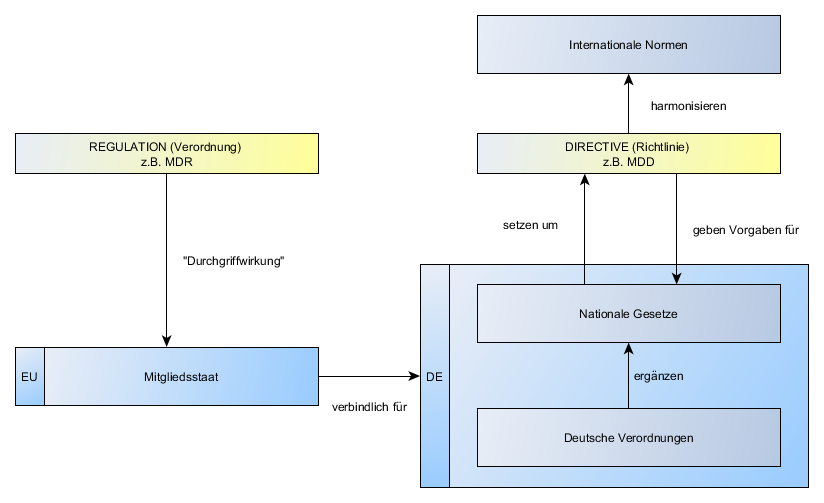
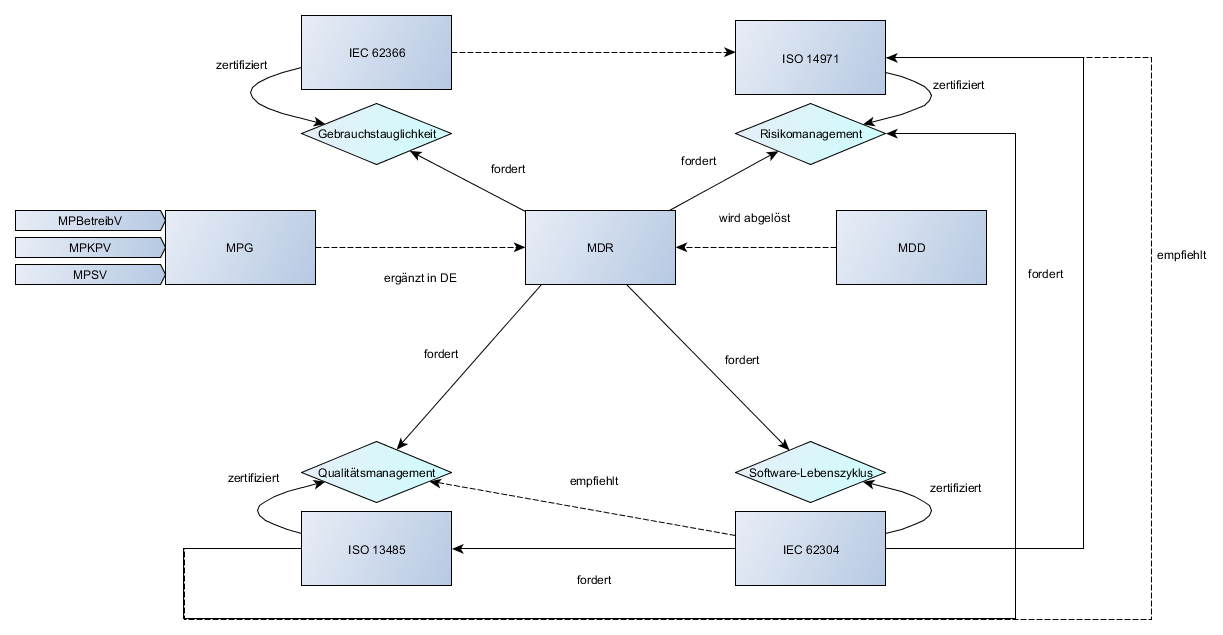
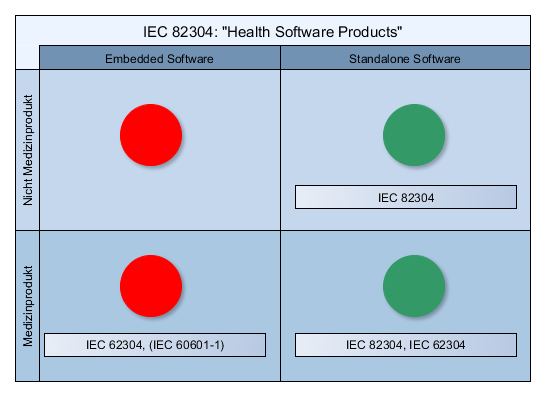
Was sind die Implant Files?
Wikipedia-Artikel zu den Implant Files: https://de.wikipedia.org/wiki/Implant_Files
Der Begriff Implant Files bezeichnet eine Ende November 2018 veröffentlichte Recherche, des Internationales Netzwerk investigativer Journalisten (ICIJ), welche weltweit Lücken in der Kontrolle von Implantaten aufdeckte.
Die Veröffentlichung erfolgte unter Zusammenarbeit von der ICIJ, BBC, WDR, NDR, Das Erste und Süddeutscher Zeitung.
Vorangegangen war eine zweijährige Recherche mit mehr als 250 Journalisten aus 36 Ländern.
Für Aufsehen sorgte die niederländische Journalistin Jet Schouten, die den ersten Schritt zur Zulassung eines Mandarinennetzes als Vaginalnetz bekommen hatte.
Problematik
Die Recherche zeigt auf, dass Medizinprodukte oftmals zu leicht zugelassen werden.
Normalerweise müssen neue Medizinprodukte eine klinische Studie durchlaufen, bevor sie zugelassen werden. Ist dies jedoch bereits mit einem ähnlichen Produkt geschehen, so gilt das Äquivalenzprinzip, und die Zulassung besteht für den Hersteller dann nur aus dem Erhalt eines CE-Zeichens, wobei der erste Schritt eine Begutachtung der technischen Dokumentation darstellt und der zweite Schritt eine Begutachtung der Fabrik des Herstellers.
Auch die Unabhängigkeit der Prüfstelle, in Deutschland z. B. die Dekra oder der TÜV, wird infrage gestellt, da der Hersteller sowohl Auftraggeber der Prüfung ist und diese ebenfalls bezahlt. Das Prüfunternehmen entscheide bei der Prüfung dann oft nur anhand der vom Hersteller bereit gestellten Unterlagen und nicht anhand des Produktes selbst.
Sollte das Produkt abgelehnt werden, so hat der Hersteller allerdings auch die Möglichkeit, das Produkt bei einem anderen von etwa 50 Prüfunternehmen prüfen zu lassen.
Außerdem wird kritisiert, dass Hersteller oft selbst Medizinprodukte zurückrufen müssen und dies nur selten behördlich angeordnet werde.
Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) ist für die Überwachung der Medizinprodukte zuständig. Es kann selbst allerdings nur Empfehlungen aussprechen und keine Verordnungen oder Verbote erlassen. Dies kann nur eine entsprechende Landesbehörde tun, die dies allerdings selten macht. Außerdem meldet ein Arzt, im Falle einer Vermutung eines Fehlers im Produkt, dies an den Hersteller. Dieser habe, einer Untersuchung des BfArM zufolge, nur in etwa der Hälfte der Fälle reagiert.
Kritisiert wird ferner, dass des keine deutschlandweite oder europaweite Datenbank über implantierte Medizinprodukte gibt, ein sogenanntes Implantatregister.
Weltweit gibt es nur in 20 % der Länder öffentliche Datenbanken mit Informationen über Sicherheitswarnungen und Rückrufe von Medizinprodukten.
Bleeding Edge
Zuvor gab es die Netflix Doku Bleeding Edge. Dort werden teilweise die gleichen Produkte behandelt.
International Medical Device Database
- https://medicaldevices.icij.org
- https://www.sueddeutsche.de/politik/implant-files-datenbanken-:patienten-1.4226388
Interessanterweise fehlen zwei interessante Länder:
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) in Deutschland fehlt
- Zulassungs- und Aufsichtsbehörde für Arzneimittel in Großbritannien MHRA fehlt
Wie sieht die Realität aus?
- https://www.rheinpfalz.de/artikel/fehlerhafte-implantate-der-inszenierte-skandal/
- https://www.bvmed.de/de/bvmed/presse/pressemeldungen/bvmed-zur-ard-doku-ausser-kontrolle-und-hart-aber-fair
- https://www.bvmed.de/de/bvmed/presse/pressemeldungen/bvmed-richtigstellungen-zur-meldung-von-vorkommnissen-mit-medizinprodukten
Schwachstellen im System
- Äquivalenzprinzip: Viele Produkte werden gar nicht erst klinisch getestet, weil bereits ähnliche Produkte auf dem Markt sind. Nur etwa 10 Prozent der Produkte in der höchsten Risikoklasse werden selbst systematisch am Patienten erprobt.
- Das Äquivalenzprinzip gibt es in Amerika. Dort können mit dem sogenannten 510(k) Verfahren “ähnliche” Produkte in Verkehr gebracht werden. Das Thema wurde in der Netflix Doku Bleeding Edge sehr anschaulich behandelt. In Europa gibt so so ein Verfahren nicht.
- Die Anforderungen an die Äquivalenz der Produkte bei der klinischen Bewertung wurden in der Vergangenheit oft großzügig ausgelegt.
- Klinische Studien: Selbst wenn der Hersteller eine Studie durchführt: die Vorgaben dafür sind lax, ein Nutzen des Produkts für den Patienten muss nicht nachgewiesen werden.
- Mit der neuen EU Leitlinie MEDDEV 2.7/1 wurden diese Anforderungen erhöht. Die benannten Stellen fordern dies auch streng ein, so streng, dass der Sinn des Äquivalenzprinzips in Frage steht. Unnötige klinischen Prüfungen nicht nur den Herstellern, sondern auch den Patienten zu ersparen. Was bewiesen ist, muss nicht nochmals bewiesen werden.
- Für diese hochkritischen Produkte führt die MDR ein sogenanntes Scrutiny-Verfahren ein.
- Benannte Stellen: Das Prüfunternehmen, das am Ende über die Zertifizierung entscheidet, prüft nicht unabhängig – denn es wird vom Hersteller beauftragt und bezahlt.
- Die Alternative zu den benannten Stellen ist eine staatliche Zulassungsstelle wie das Kraftfahrtbundesamt in Deutschland oder die FDA in den USA.
- Die FDA in Amerika prüft selbst auch keine Produkte. Das Totalversagen des Kraftfahrtbundesamts in der „Diesel-Affäre“ lässt auch meiner Sicht nicht den Schluss zu, dass staatliche Stellen besser geeignet seien.
- Prüfung der benannten Stellen: Das Prüfunternehmen entscheidet nur anhand der eingereichten Unterlagen – das Produkt selbst wird nicht untersucht.
- Diese Aussage stimmt insbesondere bei Implantaten in dieser Allgemeingültigkeit nicht. Bei Hochrisikoprodukten ist genau die Prüfung der Produktauslegung explizit gefordert und wird auch durchgeführt. Das betrifft alle Produkte der Medizinprodukteklasse III.
- Wechsel der benannten Stelle: Lehnt ein Prüfunternehmen ein Produkt ab, kann der Hersteller es einfach bei einem anderen versuchen.
- Grundsätzlich ist das erst mal korrekt.
- Die Anforderungen der neuen Medizinprodukteverordnung stellen sehr hohe Anforderungen an die benannten Stellen.
- Benannte Stellen mit zweifelhafter Kompetenz, Neutralität und Gesetzestreue wurden geschlossen.
- Hier müssen sich meiner Erachtens die benannten Stellen untereinander besser Vernetzen.
- Medizinisches Fachwissen der benannten Stellen: Es arbeiten kaum Ärzte in den benannten Stellen die den Inhalt von medizinischen Studien beurteilen können.
- Das ist leider wahr. Die benannten Stellen haben diese Problem jedoch schon vor längerem erkannt und entsprechend reagiert.
- Die benannten Stellen haben ihr Personal deutlich mit Experten wie Ärzten aufgestockt und „prüfen“ daher intensiver und kompetenter.
- Arzneimittel viel Stärker kontrolliert: Medikamente können nur auf den Markt gelangen, wenn umfangreiche Studien mit menschlichen Patienten ihre Wirksamkeit belegen. Silikonkissen oder andere risikoreiche Medizinprodukte können hingegen implantiert werden, ohne dass sie hinreichend getestet sind.
- Auch bei den Arzneimitteln gibt es schwarze Schafe
- Unterdosierte Krebsmittel
- gestohlene Krebsmittel aus Griechenland
- Blutdrucksenker mit potenziell Krebserregenden Stoff verunreinigt
- Die strengen Kontrollen bei Arzneimittel können auch nicht vor böswilligen Handeln schützen. Kein System kann das. Egal ob von einen staatlichen Behörde oder von benannten Stellen.
- Auch bei den Arzneimitteln gibt es schwarze Schafe
- Vorkommnisse: Hersteller reagieren nicht auf Probleme mit Ihren Produkten
- Es gibt mit Sicherheit Hersteller die Ihrer Pflicht nicht nachkommen
- Es ist auch korrekt, dass das BfArM hier wenig tun kann. Es hat den Eindruck das das BfArM seine Hauptaufgabe in der Veröffentlichung der Vorkommnisse der Hersteller sieht.
- Die Landesbehörden führen keine (wahrnehmbare) aktive Überwachung durch und reagieren selbst auf Anfragen zurückhaltend. Ein wirksames Überwachungssystem sieht anders aus.
- Haftpflichtversicherung: Es heißt in der neuen Verordnung nur, dass die Hersteller freiwillig eine Versicherung abschließen oder Rücklagen bilden sollen, damit sie bei Schäden durch fehlerhafte Produkte haften können.
- Hersteller werden im Audit des Qualitätsmanagementsystems (meinst nach ISO 13485) dahingehend geprüft ob eine Haftpflichtversicherung besteht.
- Das führt zu eine Hauptabweichung die der Hersteller sehr schnell beheben muss
- Eudamed Datenbank: Es ist nicht klar welche Informationen öffentlich sein werden. Außerdem scheint er Aufbau scheint aber in Verzug zu sein. Es ist spannend, ob am Ende Abstriche gemacht würden bei Aufbau, Zugang und der Art, wie Daten einpflegt werden. Wenn es nicht erreicht wird, bleibt es erst einmal beim alten, intransparenten System.
- Die Datenbank soll auch für die Öffentlichkeit (teilweise) einsehbar sein
- Es ist korrekt, dass noch nicht endgültig entschieden wurde welche Informationen öffentlich werden und welche nicht
- Dadurch wird es jedoch möglich Medizinprodukte sehr genau nachverfolgen zu können
- https://projekte.sueddeutsche.de/implantfiles/politik/implant-files-schwachstellen-im-medizinprodukte-system-e701831/
- https://www.sueddeutsche.de/politik/medizinprodukte-eu-gesetz-implant-files-1.4223924
- https://www.johner-institut.de/blog/regulatory-affairs/implant-files/
- https://www.johner-institut.de/blog/regulatory-affairs/eudamed/
Öffentlicher Pranger von Unternehmen und Personen
Die Professoren Max Aebi und Thomas Steffen:
- https://www.srf.ch/news/schweiz/implant-files-ich-kann-jederzeit-dazu-stehen
- https://www.tagesanzeiger.ch/the-implant-files/die-professoren-sollten-sich-detailliert-erklaeren/story/18082569
- https://www.tagesanzeiger.ch/the-implant-files/die-patienten-werden-zu-versuchskaninchen/story/18286624
Sie waren als Ärzte bei der klinischen Prüfung von Bandscheiben-Implantaten beteiligt waren die zu Problemen geführt haben. Es wird Ihnen Fehler bei der klinischen Prüfung vorgeworfen.
Medtronic
- https://www.tagesschau.de/inland/implantfiles/fakescience-119.html
- https://projekte.sueddeutsche.de/implantfiles/politik/implant-files-medtronic-unter-schock-e311905/
Dem Unternehmen werden Vertuschung vorgeworfen.
Bayer
War das Unternehmen, das die Essure Verhütungsspirale in Verkehr gebracht hat.
Wir hoffen, dass dadurch die Menschen nicht verunsichert werden und Angst vor medizinischen Eingriffen bekommen.
Reaktionen
Reaktionen zu den Implant Files werden sehr aktuell auf der folgenden Seite gesammelt:
Teilweise Gegendarstellung des Bayrischen Rundfunks:
Schlussworte
Die Implant Files sind gut um auf Fehler im System hinzuweisen. Eine gewisses dramatisieren ist notwendig, um aufzurütteln. Wie die Recherche gezeigt hat gibt es durchaus Medizinprodukte bei denen Grund zur Sorge besteht. Es ist unbestritten, dass jeder unnötig geschädigte Patient einer zu viel ist.
Wir hoffen, dass dadurch die Menschen nicht verunsichert werden und Angst vor medizinischen Eingriffen bekommen.
Ich persönlich halte die Relativierung der deutschen Medizintechnikverbände (BVMed, Devicemed, …) nicht für zielführend. Die gesamte Branche muss sich intensiv mit den Vorwürfen und Forderungen auseinander setzten. Da täglich neue Berichte zu den Implant Files veröffentlicht werden, müssen hier sehr viele Diskussionen geführt werden. Zum Zeitpunkt der Aufnahme konnten vom Schweizer Medizintechnikverband hat noch keine Pressemitteilung zu den Implant Files gefunden werden.
Es ist schade, dass die Veröffentlichung mit dem Wechsel der neuen Medizinprodukteverordnung so korreliert. Dadurch sind viele Forderungen der Implant Files “leicht” umzusetzen, weil sie ja schon im neuen Gesetz stehen.
Die Implant Files sind für Hersteller, benannten Stellen und Behörden eine Chance zu zeigen welche Anstrengungen unternommen werden um sichere Medizinprodukte in Verkehr zu bringen. Unser Podcast ist hier ein kleiner Beitrag um für Aufklärung zu sorgen, Informationen zu liefern und Fragestellungen zu beantworten.
Quellen
Hauptseiten der Implant Files
- https://www.icij.org/investigations/implant-files/
- https://projekte.sueddeutsche.de/implantfiles
- https://www.tagesanzeiger.ch/the-implant-files/
- https://medicaldevices.icij.org/
Aufwand für Zulassungen:
Pressemeldungen und Reaktionen:
- https://www.johner-institut.de/blog/regulatory-affairs/implant-files/
- https://www.sueddeutsche.de/politik/nachrichten-podcast-implant-files-sz-1.4225648
- https://bazonline.ch/the-implant-files/29-000-Tweets-mit-1500-ArtikelLinks-in-33-Sprachen/story/13508573
- http://www.wbur.org/hereandnow/2018/01/10/medical-devices-danger-within-us-jeanne-lenzer
- https://www.sueddeutsche.de/politik/implant-files-essure-spirale-frauen-1.4228776
- https://www.rheinpfalz.de/artikel/fehlerhafte-implantate-der-inszenierte-skandal/
- https://www.srf.ch/news/wirtschaft/fehlerhafte-implantate-medizin-firmen-geloben-besserung-im-jahr-2020
- https://www.tk.de/tk/nachgefragt-bei/im-november/medizinprodukte/991828
- https://www.tk.de/tk/pressemitteilungen/bundesweite-pressemitteilungen/991764
- https://www.devicemed.de/bv-med-richtigstellungen-zur-meldung-von-vorkommnissen-mit-medizinprodukten-a-780569/
- https://gmp-podcast.de/blog/gmp037/
- https://www.bvmed.de/de/bvmed/presse/pressemeldungen/bvmed-befuerwortet-implantateregister
- https://www.bvmed.de/de/bvmed/presse/pressemeldungen/bvmed-zur-ard-doku-ausser-kontrolle-und-hart-aber-fair
- https://www.bvmed.de/de/bvmed/presse/pressemeldungen/bvmed-zu-implantfiles