Grundlagen EKG
- Elektrokardiogramme, häufig auch als EKG bezeichnet, zeichnen den zeitlichen und örtlichen Verlauf elektrischer Erregungsvorgänge am Herzmuskel auf
- Jede Erregungsausbreitung über Fasern im Herzen stellt eine Quelle elektrischer Spannung dar
- Das Herz ist somit eine Potentialquelle und das EKG zeigt quasi ein Bild der Potentialverschiebung oder Elektrizitätserzeugung, also die Erregungsvorgänge am Herzen
- Bei einem klassischen Elektrokardiogramm liegt die Größe des Nutzsignals bei ca. einem Millivolt.
- Unterschiede finden sich in der Abnahmetechnik und in räumlicher Hinsicht
- Als bipolare Ableitung bezeichnet man Ableitungen zwischen zwei Punkten
- Bei der so genannten unipolaren Ableitung ist eine differente Elektrode gegen eine Nullelektrode geschaltet
- Als Standardableitungen werden bezeichnet: Die drei bipolaren Extremitätenableitungen (I, II, III) nach Einthoven, die auf diesen Ableitungen basierenden und von Goldberger konstruierten unipolaren Extremitätenableitungen (aVR, aVL, aVF), sowie die sechs unipolaren Brustwandableitungen (V1, V1, V3, V4, V5, V6) nach Wilson
- Ein konventionelles 12-Kanal-EKG registriert parallel die Extremitätenableitungen nach Einthoven (3) und Goldberger (3), sowie die Brustwandableitung nach Wilson (6)
Der Artikel in der Wikipedia beschreibt das Thema EKG auch sehr gut. Dort werden auch die verschiedenen Abschnitte eines EKGs erklärt.
Quellen:
- R. Kramme (Hrsg.), Medizintechnik, 4. Auflage, Springer-Verlag Berlin Heidelberg 2011
- https://flexikon.doccheck.com/de/Elektrokardiogramm
Apple Watch und EKG
Die Apple Watch besitzt ein sog. 1-Kanal EKG. Generell gilt für 1-Kanal EKGs das Folgende:
- ein 1-Kanal EKG ist eine unipolare Ableitung nach Wilson
- es werden eine oder zwei Elektroden verwendet
- es können nur Rythmusanalysen durchgeführt werden
- 1-Kanal EKGs können nicht zur kardiologischen Diagnose verwendet werden
- Ischämien können überhaupt nicht erkannt werden und auch die Aussagekraft für Diagnose und Lokalisierung von Herzrhythmusstörungen ist sehr stark eingeschränkt
- das 1-Kanal-EKG dient in erster Linie der Erkennung von Tachykardien und (häufiger) der von Bradykardien
- es gibt 1-Kanal-EKGs für Hausärzte die auf die Brust des Patienten gelegt werden können
- 1-Kanal EKG Produkte: (Zufällig gefunden ohne Anspruch auf Vollständigkeit)
Aus dem 1-Kanal-EKG lassen sich aber nur folgende Diagnosen stellen:
- Bradykardie (zu langsamer Herzrhythmus)
- Tachykardie (zu schneller Herzrhythmus)
- Asystolie (kein Rhythmus)
- supraventrikuläre und ventrikuläre Extrasystolen (Herzschläge außerhalb des physiologischen Herzrythmus)
- ektoper Rhythmus (eine Art von Herzrhythmusstörung)
- Vorhof- und Kammerflattern oder -flimmern
Apple hatte Mitte 2016 in den USA ein Patent für eine EKG-Funktion in einem Wearable angemeldet:
Quelle: R. Kramme (Hrsg.), Medizintechnik, 4. Auflage, Springer-Verlag Berlin Heidelberg 2011
“Konkurrenzprodukte” unter anderem von:
1. Alivecor
Firma: https://www.alivecor.com/
Gebrauchsanweisung: https://www.alivecor.com/previous-labeling/kardia/08LB12.5.pdf
FDA: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm
In der Suche bei Applicant Name nach alivecor suchen: Hier sieht man, es wurde sieben mal eine 510(k), eine Art “Erlaubnis”, ausgestellt.
- der Hersteller hat die Konformität in der EU erklärt (CE-Zeichen)
- benannte Stelle war der TÜV Süd (0123) auf dem CE-Zeichen in der Gebrauchsanweisung
Zweckbestimmung:
Das Produkt Kardia Mobile ist für die Aufzeichnung, Speicherung und Übertragung eines Einkanaligen Elektrokardiogramm (EKG) Rhythmus bestimmt. Das Produkt Kardia Mobile zeigt auch EKG-Rhythmen an und erkennt das Vorhandensein von Vorhofflimmern und normalem Sinusrhythmus (wenn verschrieben oder unter ärztlicher Aufsicht verwendet). Das Produkt Kardia Mobile ist für den Gebrauch durch medizinisches Fachpersonal, Patienten mit bekannten oder vermuteten Herzerkrankungen und gesundheitsbewusste Personen bestimmt. Das Produkt ist nicht für den pädiatrischen Einsatz vorgesehen und wurde auch nicht dementsprechend getestet.
2. Personal MedSystems GmbH
Firma: https://www.cardiosecur.com/de/produkt/wie-es-funktioniert/
Gebrauchsanweisung: https://www.cardiosecur.com/de/meta-navigation/footer/hilfe/download-center/
- der Hersteller hat die Konformität in der EU erklärt (CE-Zeichen)
- benannte Stelle war der TÜV Süd (0123) auf dem CE-Zeichen in der IFU
Zweckbestimmung
CardioSecur dient dazu, Ruhe-Elektrokardiogramme (-EKGs) von Erwachsenen aufzuzeichnen, auszuwerten und zu speichern. CardioSecur ist ein medizinisches, elektrisches System, welches aus einem EKG-Kabel und einer App für das Smartphone besteht. Mit 4 Elektroden kann CardioSecur ein vollwertiges 15-Kanal-EKG aufzeichnen, auswerten und speichern. Anhand des Vergleiches eines Kontroll-EKGs mit einem Referenz-EKG erkennt CardioSecur mögliche Rhythmus- und Durchblutungsstörungen des Herzens. CardioSecur dient der Selbstüberwachung der Herzaktivität.
Die Aufzeichnung des Referenz-EKGs muss zu einem beschwerdefreien Zeitpunkt erfolgen und wird in der CardioSecur App gespeichert. Die Aufzeichnung kann sowohl unter ärztlicher Kontrolle als auch durch den Anwender selbst erfolgen. Ein Kontroll-EKG sollte sofort bei Beschwerden oder aber unabhängig von Beschwerden in regelmäßigen Abständen (z. B. täglich) erfolgen.
Der Anwender benötigt für die Verwendung von CardioSecur keine medizinischen Vorkenntnisse und muss nicht gesondert geschult sein. CardioSecur wird für Personen mit einer bekannten oder vermuteten Herzerkrankung und für Personen mit Risikofaktoren für eine Herzerkrankung (z. B. Bluthochdruck, Diabetes mellitus oder erhöhtes Cholesterin) empfohlen. CardioSecur kann sowohl von Patienten als auch von Vorsorgenden verwendet werden. […]
Was ist an der Apple Watch ein Medizinprodukt?
Kurze Wiederholung der Definition Medizinprodukt
- Artikel 1 MDD: https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:01993L0042-20071011&from=EN
- Artikel 2 MDR: https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:32017R0745&from=DE#d1e1258-1-1
Sehr starkt vereinfacht zusammengefasst: Medizinprodukte sind Produkte, die vom Hersteller dazu bestimmt sind
- Krankheiten und Verletzungen von Patienten zu diagnostizieren, zu überwachen und zu therapieren oder
- physiologische Vorgänge zu untersuchen oder
- den anatomischen Aufbau von Menschen zu verändern oder
- der Empfängnisregelung zu dienen.
Sobald ein Produkt einer dieser Definitionen genügt, ist es ein Medizinprodukt, das den regulatorischen Anforderungen genügen muss. Das bedeutet, dass der Hersteller (mit seiner Zweckbestimmung) entscheidet, ob es sich bei dem Produkt um ein Medizinprodukt handelt.
Wie ist das jetzt mit der “Zulassung” in Amerika von der FDA?
Die FDA hat für zwei Apps auf der Uhr eine “Clearance” (Freigabe) erteilt. Das ist nicht das gleiche wie ein “Approval” (Zulassung”). Approvals gibt es von der FDA nur für Klasse III Produkte (z.B.: Implantierbare Herzschrittmacher).
Das aufwändigste Verfahren ist die FDA-Zulassung, die nur für Produkte der Klasse III erfolgt, oder für Technologien, die ein höheres Risiko, aber auch einen höheren Nutzen haben könnten. https://www.fda.gov/medicaldevices/productsandmedicalprocedures/deviceapprovalsandclearances/default.htm
Apple hat eine “de novo”-Klassifizierung für die beiden Apps erhalten. Das bedeutet,
- die Apps liegen risikobedingt noch in der Klasse II
- die Apps haben nicht so viele Tests durchlaufen hat wie ein “zugelassenes” Gerät
- die Apps sind anders als alles andere auf dem Markt (de novo Charakter)
Interessante Infos, bevor wir in die FDA Unterlagen schauen
Die ehemalige FDA Beamtin Donna-Bea Tillman hat die Einreichung bei der FDA für Apple durchgeführt. Da hat Apple eine exzellente Wahl getroffen.
- https://twitter.com/MedtechDavid/status/1039944765893226498
- https://www.biologicsconsulting.com/consultant-bio/donna-bea-tillman/
DeNovo Datenbank: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/denovo.cfm
- bei Requester Name nach Apple suchen
EKG App
FDA Dokumente zur de novo Klassifizierung: https://www.accessdata.fda.gov/cdrh_docs/pdf18/DEN180044.pdf
- Device Class: 2
- Product Code: QDA
Zweckbestimmung aus der DeNovo Klassifizierung
Die EKG-App ist eine reine mobile medizinische Softwareanwendung, die für die Verwendung mit der Apple Watch bestimmt ist, um ein Einkanal-EKG zu erstellen, aufzuzeichnen, zu speichern, zu übertragen und anzuzeigen, ähnlich dem eines 1-Kanal EKGs. Die EKG-App bestimmt das Vorhandensein von Vorhofflimmern (AFib) oder Sinusrhythmus auf einer klassifizierbaren Wellenform. Die EKG-App wird nicht für Benutzer mit anderen bekannten Arrhythmien empfohlen.
Die EKG-App ist für den rezeptfreien Einsatz (OTC) vorgesehen. Die von der EKG-App angezeigten EKG-Daten sind nur zur Information bestimmt. Der Benutzer ist nicht dazu bestimmt, ohne Rücksprache mit einem qualifizierten medizinischen Fachpersonal klinische Maßnahmen auf der Grundlage der Geräteausgabe zu interpretieren oder zu ergreifen. Die EKG-Wellenform soll die Rhythmusklassifizierung ergänzen, um AFib vom normalen Sinusrhythmus zu unterscheiden, und nicht dazu bestimmt, traditionelle Diagnose- oder Behandlungsmethoden zu ersetzen. Die EKG-App ist nicht für Personen unter 22 Jahren bestimmt.
Die FDA identifiziert diesen generischen Gerätetyp als elektrokardiographische Software für den rezeptfreien Gebrauch. Eine elektrokardiographische Gerätesoftware für den rezeptfreien Gebrauch erzeugt, analysiert und zeigt elektrokardiographische Daten an und kann Informationen zur Identifizierung von Herzrhythmusstörungen liefern. Dieses Gerät ist nicht dazu bestimmt, eine Diagnose zu stellen.
Irregular Rhythm Notification Feature
FDA Dokumente zur de novo Klassifizierung: https://www.accessdata.fda.gov/cdrh_docs/pdf18/DEN180042.pdf
- Device Class: 2
- Product Code: QDB
Zweckbestimmung aus der DeNovo Klassifizierung
Das Irregular Rhythm Notification Feature ist eine reine mobile medizinische Softwareanwendung, die für die Verwendung mit der Apple Watch bestimmt ist. Die Funktion analysiert Pulsfrequenzdaten, um Abschnitte unregelmäßiger Herzrhythmen zu identifizieren, die auf Vorhofflimmern (AFib) hinweisen und informiert den Anwender. Die Funktion ist für den rezeptfreien Einsatz (OTC) vorgesehen. Es ist nicht vorgesehen, über jeden Abschnitt eines unregelmäßigen Rhythmus, der auf AFib hindeutet, zu informieren, und das Fehlen einer Benachrichtigung soll nicht darauf hindeuten, dass kein Erkrankungsverlauf vorliegt; vielmehr soll das Merkmal opportunistisch eine Benachrichtigung über eine mögliche AFib hervorrufen, wenn ausreichende Daten für die Analyse verfügbar sind. Diese Daten werden nur erfasst, wenn sich der Benutzer noch im Ruhezustand befindet. Zusammen mit den Risikofaktoren des Benutzers kann die Funktion genutzt werden, um die Entscheidung für das AFib-Screening zu ergänzen. Das Merkmal ist nicht als Ersatz für herkömmliche Diagnose- oder Behandlungsmethoden gedacht.
Die Funktion wurde nicht getestet und ist nicht für die Verwendung bei Personen unter 22 Jahren vorgesehen. Es ist auch nicht für die Verwendung bei Personen bestimmt, bei denen zuvor AFib diagnostiziert wurde.
Die FDA identifiziert diesen generischen Gerätetyp als photoplethysmographische Analysesoftware für den stationären Einsatz. Eine Photoplethysmographie-Analyse-Softwarevorrichtung für den rezeptfreien Gebrauch analysiert Photoplethysmographie-Daten und liefert Informationen zur Identifizierung unregelmäßiger Herzrhythmen. Dieses Gerät ist nicht dazu bestimmt, eine Diagnose zu stellen.
Wie sieht es in Europa (und damit auch Deutschland) aus?
In Europa und damit auch in Deutschland sind die beiden Apps nicht verfügbar, da Apple keine Konformität nach EU-Recht erklärt hat.
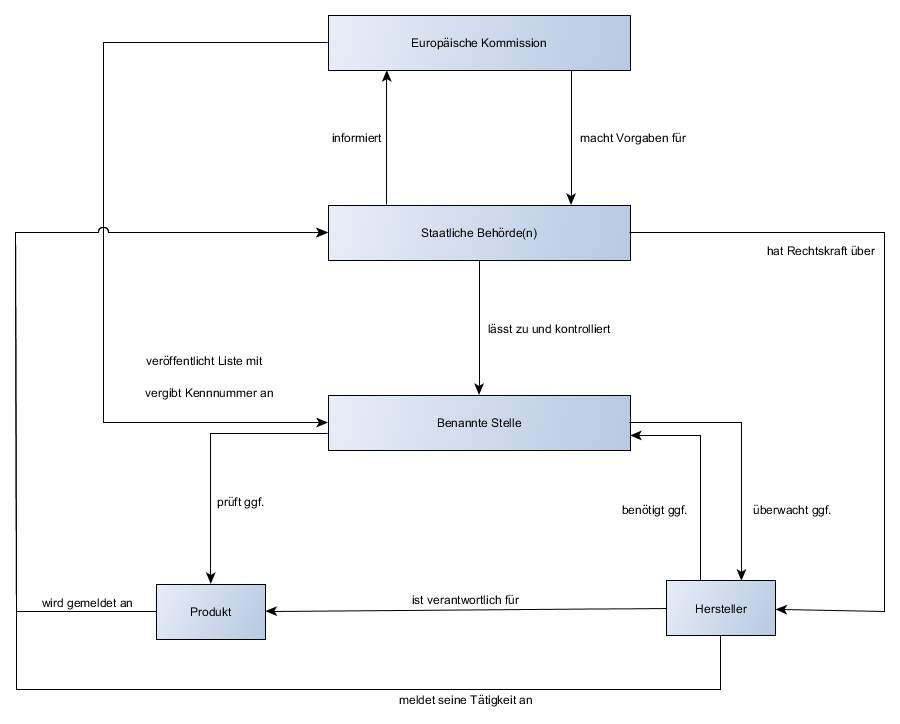
In Europa erklären die Hersteller die Konformität selbst. Eine benannte Stelle ist meist in das Konformitätsbewertungsverfahren eingebunden.
Es wird sich vermutlich um ein aktives Medizinprodukt handeln.
- Regel 10 MDD
- Regel 9, 10, 11 MDR
Auszug Regel 10 MDD:
Alle aktiven diagnostischen Produkte gehören zur Klasse IIa,
- wenn sie dazu bestimmt sind, eine direkte Diagnose oder Kontrolle von vitalen Körperfunktionen zu ermöglichen, es sei denn, sie sind speziell für die Kontrolle von vitalen physiologischen Parametern bestimmt und die Art der Änderung dieser Parameter könnte zu einer unmittelbaren Gefahr für den Patienten führen, z.B. Änderung der Herzfunktion, der Atmung oder der Aktivität des zentralen Nervensystems, oder wenn sie für die Diagnose in klinischen Situationen, in denen der Patient in unmittelbarer Gefahr schwebt, bestimmt sind; in diesen Fällen werden sie der Klasse IIb zugeordnet
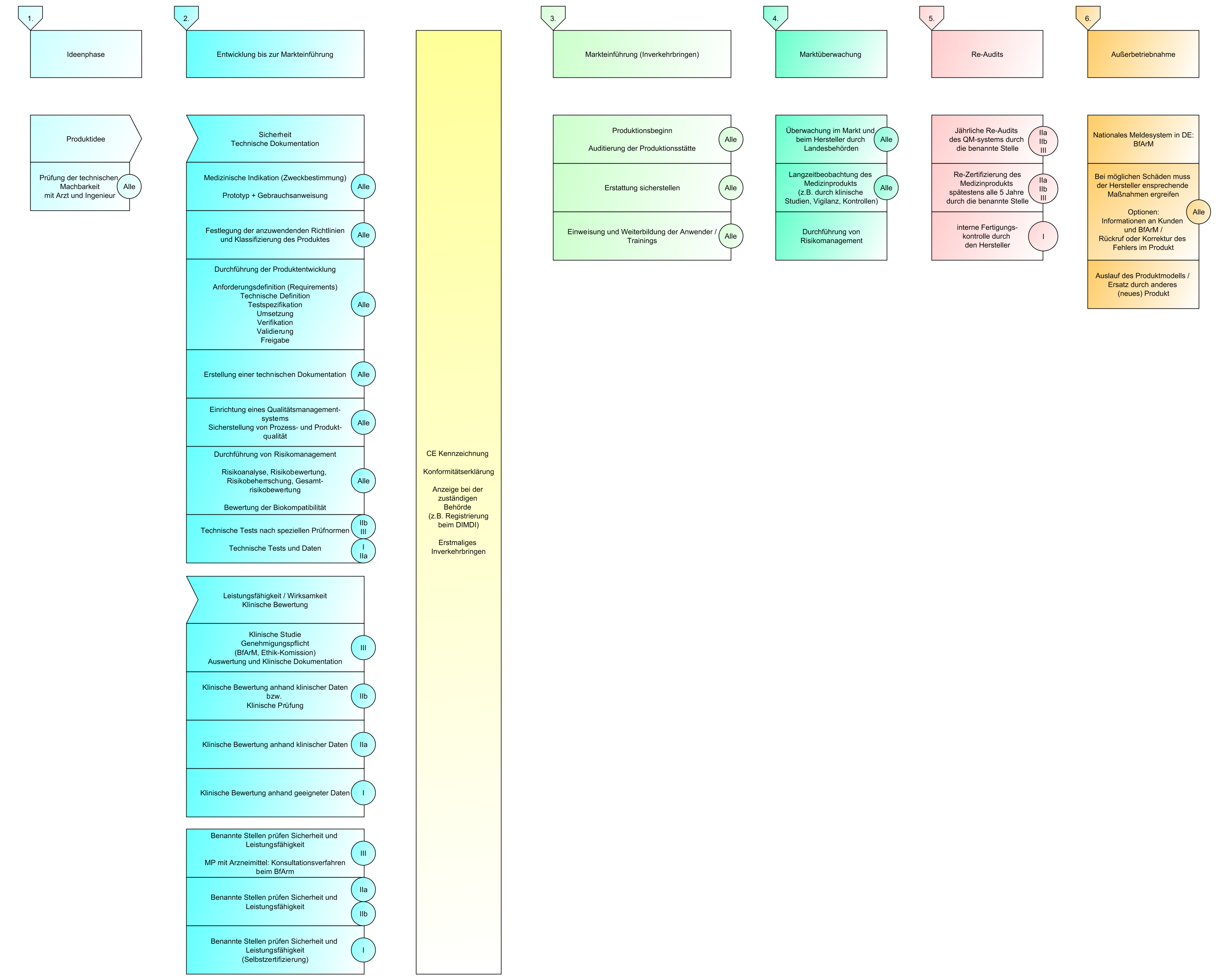
Siehe Episoden MST-002 (Rechtliche Grundlagen MDR) und MST-003 (Anwendung rechtlicher Grundlagen)